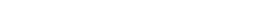
Guedes, I. A. et al. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci Rep 11, 5543 (2021).
Welcome to DockThor
A Free Web Server for Protein-ligand Docking
Protein
Add missing hydrogen atoms, complete side chains, change protonation states. Simple and easy!
Small molecules
Add hydrogen atoms (pH 7), freeze rotatable bonds, get MMFF94S atom types and partial charges. Fast and automatic!
Cofactors
Consider cofactors and structural waters on virtual screening experiments with automatic MMFF94S parametrization.
Redocking
Validate docking protocol with redocking experiments. We provide the RMSD between reference and docked poses.
Blind Docking
Searching for binding sites? Perform blind docking on the entire protein and find cavities!
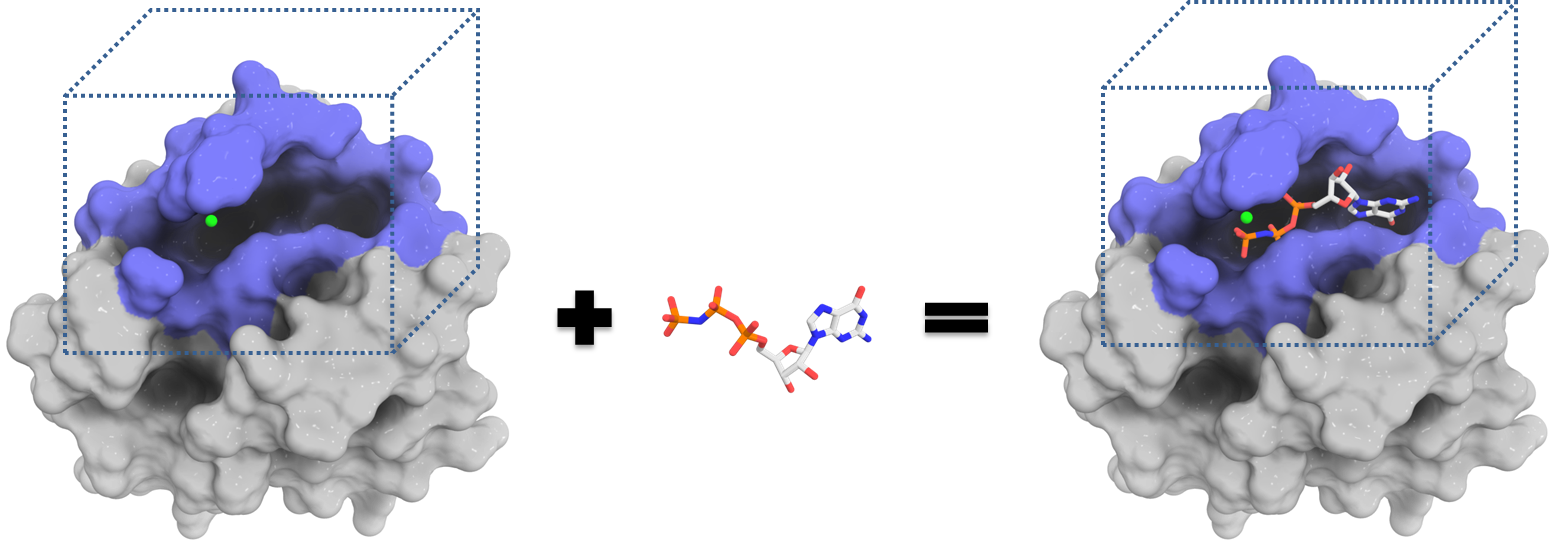
Virtual Screening
Perform large scale docking experiments exploring multiple binding modes. Dock them all!
Interactive Analyses. Explore docking poses and predict affinity.
Investigate different binding modes and predict binding affinities. Visualize predicted complexes with JSMol.

SDumont Supercomputer. Virtual screening experiments even faster.
Run virtual screening experiments at Santos Dumont supercomputer, located at LNCC, Petrópolis - Brazil.
-
Upload your protein file
-
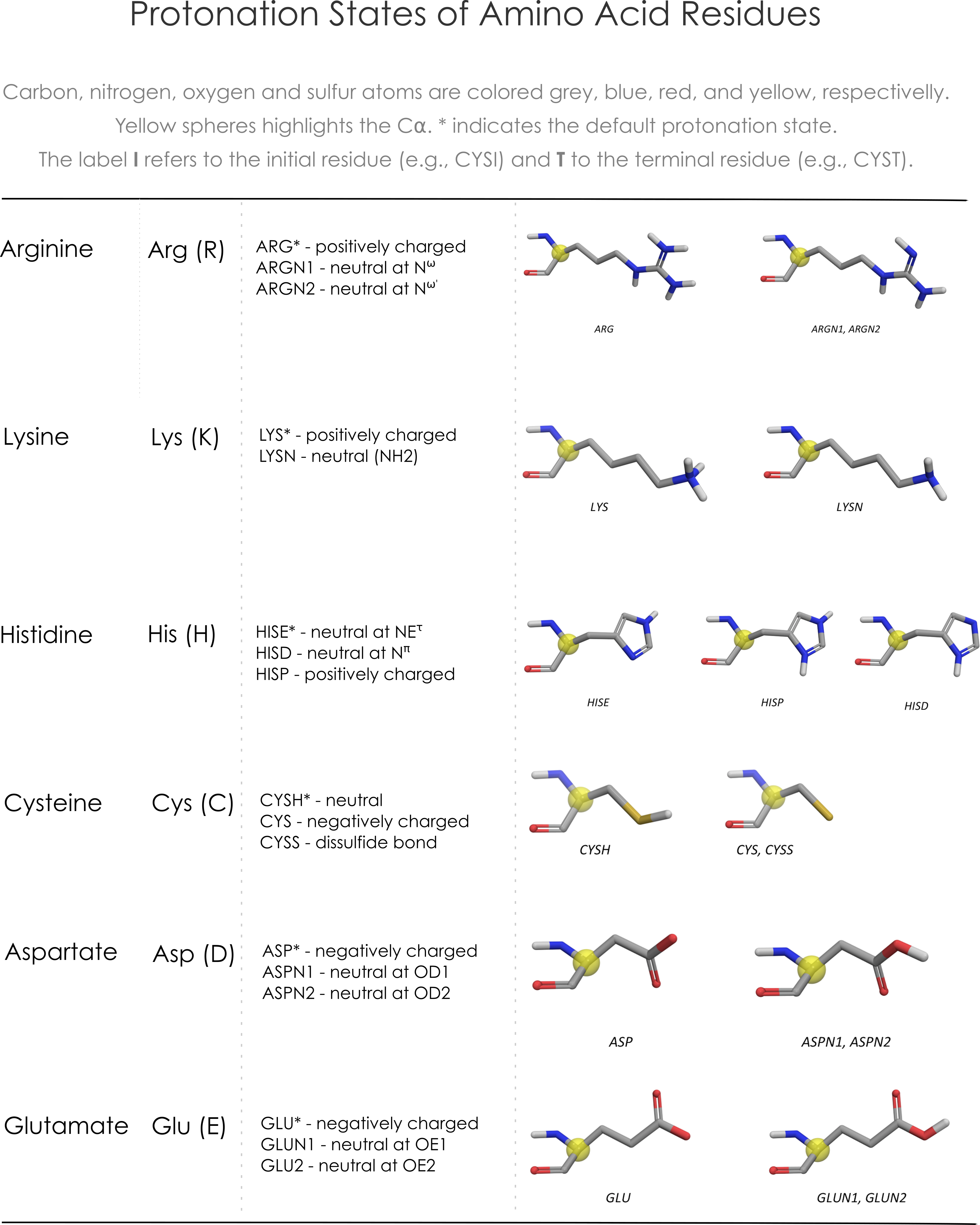
Select the protonation states
Chain {{ chainKey }}-
{{ atomKey }}
Residue index Residue number Protonation state {{indexKey}} {{indexValue.state.index}} -
* This area is just avaliable with 'pdb' files
Prepared protein file
-
Upload your ligand file
-
Rotatable bond editor
* This area is just avaliable when working with one valid ligand (pdb, sdf or mol2)
-
Prepared ligand file
-
Upload your cofactor file
-
Prepared cofactor file
-
Check your docking input files
-
Protein ( 1 )
- {{$parent.proteinInput.codedName}}
-
Ligand ( {{getLigandTotalStructures()}} )
- {{ligand.fileIdWithExtension}} ( {{ligand.validStructure}} )
-
Cofactor ( {{getCofactorTotalStructures()}} )
- {{cofactor.fileIdWithExtension}} ( {{cofactor.validStructure}} )
-
-
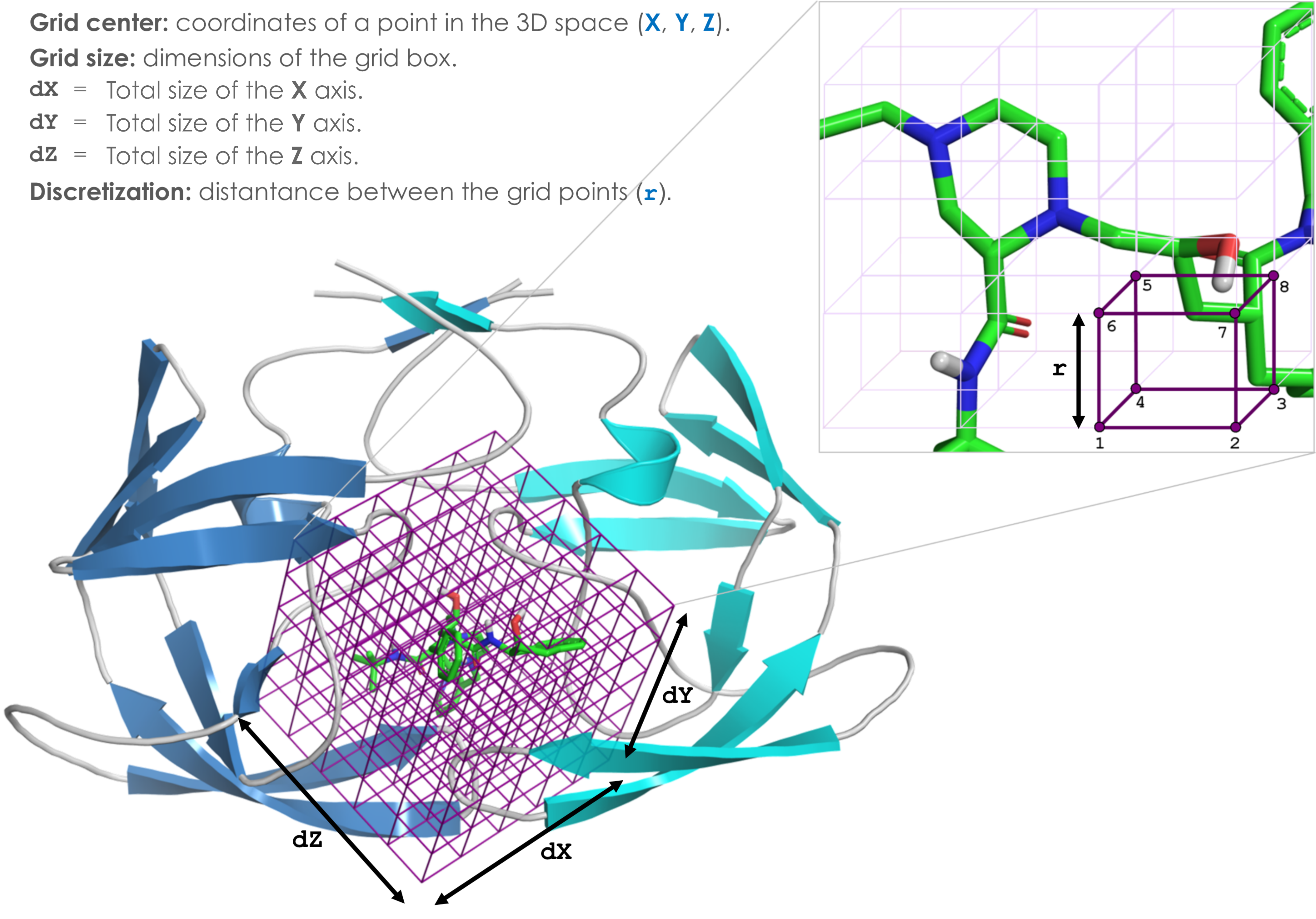
Define the binding site
Attention: the current version of DockThor (released on April 17, 2020) requires the total size of the grid box instead of the half value on each dimension. For example, now the input grid size for the docking with the test files are X = 20, Y = 20 and Z = 20 instead of 10 Å on each axis.Grid center:
X:
Y:
Z:
Grid size:
X:
Y:
Z:
Discretization:
Total grid points:
{{points}}Protein
Cofactors
-
Select the search algorithm precision
-
Identify your docking job
(success)(success)
- less than 10000 atoms;
- less than 1000 amino acid residues;
- only contains standard amino acid residues (e.g. MSE is not recognized);
- only three-dimensional structures are accepted;
- the initial amino acid must contain all atoms of the backbone.
- smaller than or equal to 10Mb;
- must contain less than 1000 atoms;
- all atoms are recognized by the MMFF94 force field;
- only three-dimensional structures are accepted.
- grid size must be larger than the ligand size;
- protein-cofactor complexes with too many atoms may fail (e.g. protein-DNA complexes);
- carefully check your email address.
The docking job {{job.id}} has failed. Please check the following requirements:
Protein file:
Ligand and cofactor files:
Docking parameters and submission:
-
Job Status
Job id "{{job.id}}" not foundJob id: {{job.id}}Job Status: {{job.status}}
-
{{ atomKey }}
References
Citing Us
- Guedes, I. A.; Pereira da Silva, M. M.; Galheigo, M.; Krempser, E.; de Magalhães, C. S.; Correa Barbosa, H. J.; Dardenne, L. E. DockThor-VS: A Free Platform for Receptor-Ligand Virtual Screening. Journal of Molecular Biology 2024, 168548. https://doi.org/10.1016/j.jmb.2024.168548.
-
Guedes, I. A.; Barreto, A. M. S.; Marinho, D.; Krempser, E.; Kuenemann, M. A.; Sperandio, O.; Dardenne, L. E.; Miteva, M. A. New Machine Learning and Physics-Based Scoring Functions for Drug Discovery. Sci Rep 2021, 11 (1), 3198. https://doi.org/10.1038/s41598-021-82410-1.
Download the curated PDBbind core set v.2013 data set here -
K. B. dos Santos, I. A. Guedes, A. L. M. Karl, and L. Dardenne. Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein-peptide Dataset, J. Chem. Inf. Model., Jan. 2020, doi: 10.1021/acs.jcim.9b00905.
Download the LEADS-PEP data set here - C. S. de Magalhães, D. M. Almeida, H. J. C. Barbosa, and L. E. Dardenne, A dynamic niching genetic algorithm strategy for docking highly flexible ligands, Information Sciences, vol. 289, pp. 206–224, Dec. 2014, doi: 10.1016/j.ins.2014.08.002.
- Guedes, I. A. et al. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci Rep 11, 5543 (2021).
When using any SARS-CoV-2 target or drug repurposing datasets, please cite the following article:
Other Articles
- I. A. Guedes, F. S. S. Pereira, and L. E. Dardenne, Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges, Front. Pharmacol., vol. 9, pp. 1–18, 2018, doi: 10.3389/fphar.2018.01089.
- I. A. Guedes, C. S. de Magalhães, and L. E. Dardenne, Receptor–ligand molecular docking, Biophysical Reviews, vol. 6, no. 1, pp. 75–87, Mar. 2014, doi: 10.1007/s12551-013-0130-2.
- C. S. de Magalhães, H. J. C. Barbosa, and L. E. Dardenne, Selection-Insertion Schemes in Genetic Algorithms for the Flexible Ligand Docking Problem, in Genetic and Evolutionary Computation – GECCO 2004, vol. 3102, K. Deb, Ed. Berlin, Heidelberg: Springer Berlin Heidelberg, 2004, pp. 368–379.
- C. S. de de Magalhães, H. J. C. Barbosa, and L. E. Dardenne, A genetic algorithm for the ligand-protein docking problem, Genetics and Molecular Biology, vol. 27, no. 4, pp. 605–610, 2004, doi: 10.1590/S1415-47572004000400022.
Thesis
- Isabella Alvim Guedes. Development of Empirical Scoring Functions for Predicting Protein-ligand Binding Affinity. D.Sc. Thesis, Laboratório Nacional de Computação Científica/Brasil, 2016.
- Diogo Marinho Almeida. Dockthor: Implementação, Aprimoramenteo e Validação de um Programa de Docking Receptor-Ligante. M.Sc. Thesis, Laboratório Nacional de Computação Científica/Brasil, 2011.
- Camila Silva de Magalhães. Algoritmos Genéticos para o Problema de Docking Proteína-Ligante. D.Sc. Thesis, Laboratório Nacional de Computação Científica/Brasil, 2006.
DockThor Team

Laurent Emmanuel Dardenne
Dr.Sci. in Biophysics
Grupo de Modelagem Molecular de Sistemas Biológicos
Laboratório Nacional de Computação Científica (LNCC-MCTI)
E-mail: dardenne@lncc.br

Hélio José C. Barbosa
D.Sc. in Civil Engineering
Grupo de Metaheurísticas Inspiradas na Natureza e Aplicações
Laboratório Nacional de Computação Científica (LNCC-MCTI)
E-mail: hcbm@lncc.br

Camila Silva de Magalhães
D.Sc. in Computational Modelling
Universidade Federal do Rio de Janeiro (UFRJ)
E-mail: camila.mag@gmail.com

Diogo Marinho Almeida
D.Sc. Student in Biotecnology
Universidade Federal do Pará (UFPA)
E-mail: diogo.marinho@gmail.com

Eduardo Krempser da Silva
D.Sc. in Computational Modelling
Grupo de Metaheurísticas Inspiradas na Natureza e Aplicações
Fundação Oswaldo Cruz (Fiocruz)
E-mail: eduardo.krempser@fiocruz.br

Fábio Lima Custódio
Dr.Sci. in Computational Modeling
Grupo de Modelagem Molecular de Sistemas Biológicos
Laboratório Nacional de Computação Científica (LNCC-MCTI)
E-mail: flc@lncc.br

Isabella Alvim Guedes
D.Sc. in Computational Modelling
Grupo de Modelagem Molecular de Sistemas Biológicos
Laboratório Nacional de Computação Científica (LNCC-MCTI)
E-mail: isabella@lncc.br
SINAPAD Team

Antônio Tadeu Azevedo Gomes
Dr.Sc. in Computer Science
Laboratório Nacional de Computação Científica (LNCC-MCTI)
E-mail: atagomes@lncc.br

Marcelo Monteiro Galheigo
Technologist in Information and Communication Technology
Laboratório Nacional de Computação Científica (LNCC-MCTI)
E-mail: galheigo@lncc.br

Vívian Medeiros
M.Sc. in Computational Modelling
Laboratório Nacional de Computação Científica (LNCC-MCTI)
E-mail: vivian@lncc.br
Contact Us
New Features
General
- 04/27/2020 - Three-dimensional structures now are visualized with the NGL Viewer.
- 04/10/2020 - New redocking Tutorial with new test files available (download the tutorial at Support -> Help).
- 01/24/2020 - Download of the curated LEADS-PEP data set is available in the References tab.
Protein
- 26/01/2024 - Modelled structure of human TMPRSS2 (template PDB code 6t7p) available in the dataset of COVID-related targets.
- 06/08/2020 - Curated structures of relevant therapeutic targets of SARS-CoV-2 are now available. The dataset will be constantly updated with new structures (wild type and selected mutations).
- 04/27/2020 - Neutral cisteine (CYSH) set as the default protonation state.
- Hydrogen atoms added to the protein with external tools (e.g. Maestro, Protoss, PDB2PQR) are automatically recognized and the residues protonation states are identified. Residues protonation states could also be modified later.
-
Download options:
- Prepared file (.pdb) containing the prepared protein file with the final protonation states and polar hydrogen atoms added.
- Topology file(s) (.in) for the uploaded target protein.
- Compacted folder (.zip) with all input and output files.
- Bug fixes.
Cofactors
- Upload up to 10 cofactor files.
- MOL2 and SDF format files are now accepted in addition to the PDB and TOP types.
- Download options:
- Map file (.csv) containing the relationship between the original file names and random IDs provided by DockThor.
- Topology file(s) (.top) for the uploaded compounds.
- Compacted folder (.zip) with all input and output files.
- Bug fixes.
Ligands
- 01/08/2024 - Virtual screening operating at reduced capacity temporarily: up to 100 small molecules as guest user and up to 1,000 compounds as registered user.
- 29/01/2024 - Virtual screening with up to 1,000 small molecules as guest user.
- 29/01/2024 - Virtual screening now with up to 10,000 small molecules as registered user.
- 26/01/2024 - Curated e-Drug3D (version June 2023) datasets for drug repurposing at reference, low and high pH are now available.
- 08/12/2020 - Curated datasets for drug repurposing at reference pH are now available! New datasets will be available soon.
- 08/12/2020 - Virtual screening with up to 200 small molecules as guest user.
- 05/22/2020 - Virtual screening now with up to 5000 small molecules as registered user.
- 01/17/2020 - Dock highly flexible ligands such as peptides with up to 40 residues and 60 dihedrals.
- Upload single or multiple structures file (only available for MOL2 and SDF files).
- Compounds are filtered to remove invalid structures:
- Molecular weight > 1500 Da;
- Rotatable bonds > 60;
- 2D structure;
- At least one atom type not recognized by the MMFF94S force field.
- Recovery the original file names with the table mapfile.csv.
- Enable/disable all rotatable bonds with just one click.
- Calculate compound properties with Obprop/OpenBabel (provided in table obprop.csv).
- Download options:
- Map file (.csv) containing the relationship between the original file names and random IDs provided by DockThor.
- Properties table (s) (.csv) containing the compounds properties calculated by Obprop/OpenBabel.
- Topology file(s) (.top) for the uploaded compounds.
- Compacted folder (.zip) with all input and output files.
- Bug fixes.
Docking
- 04/27/2020 - The input grid size on each dimension corresponds now to the total size of the grid box on each axis (instead of the half value).
- 04/10/2020 - Blind docking option that centers the energy grid at the center of mass of the protein and defines the grid dimensions to cover the entire protein (or the maximum size allowed). Attention: experimental, use it with caution.
- 01/24/2020 - Softening the MMFF94S Buf-14-7 potential now available (buffering constant = 0.35).
- Faster virtual screening with the Santos Dumont supercomputer:
- Minutes for screening up to 200 compounds;
- Few hours for screening up to 5000 molecules.
- Summary of uploaded files.
- Iterative configuration of the grid integrated with the protein visualization.
- Pre-defined configurations of the search algorithm:
- Standard: optimized configuration explored through in-house benchmarking studies.
- Virtual screening: faster protocol for screening compound databases with good accuracy.
- Explorer: change the parameters as you wish (only available for docking with up to 100 compounds).
- Receive the e-mail with the results in up to five addresses.
- Subscribe DockThor e-Newsletters to receive DockThor news.
- Job name simplified to label + random ID.
- An e-mail is sent when your job is submitted and a second one when the job is finished.
Analyses
- 04/27/2020 - Number of top-energy binding modes are limited to 3 for virtual screening and 10 for single docking experiments.
- 04/10/2020 - Corrected the protein file *_prep.pdb in the docking compacted folder (.zip file) at the directory PROTEIN/.
- Predict binding affinity with a new empirical scoring function composed by terms accounting for intermolecular interactions, ligand entropy, desolvation and lipophilic contacts.
- Explore multiple binding modes even in virtual screening experiments.
- Download your docking results:
- Ranked compounds according to the affinity prediction (bestranking files).
- Top-ranked binding modes for each compound after clustering (result- files).
- Explore different clustering parameters when docking a single ligand.
Releases Notes
- Camila Silva de Magalhães, Diogo Marinho Almeida, Hélio José Correa Barbosa, Laurent Emmnanuel Dardenne. A Dynamic Niching Genetic Algorithm Strategy for Docking of Highly Flexible Ligands. Information Sciences, 289, p. 206-224, 2014.
- Camila Silva de Magalhães, Hélio José Correa Barbosa, Laurent Emmnanuel Dardenne. Selection-Insertion Schemes in Genetic Algorithms for the Flexible Ligand Docking Problem. Lecture Notes in Computer Science, v. 3102/2004, p. 1-12, 2004.
Why to register?
- Submit virtual screening experiments with up to 5000 ligands.
- Perform virtual screening experiments with the Santos Dumont supercomputer (according to availability).
- Run your job with priority.
How to get a login?
- Submit a short research project following the instructions.
- The project will be evaluated by an internal committee.
- In the case of the acceptance of your proposal, you will receive an e-mail with the instructions to access the restrict area of the DockThor web server.


Grid box parameters
Version 2.0 . Copyright © GMMSB 2019. All Rights Reserved.